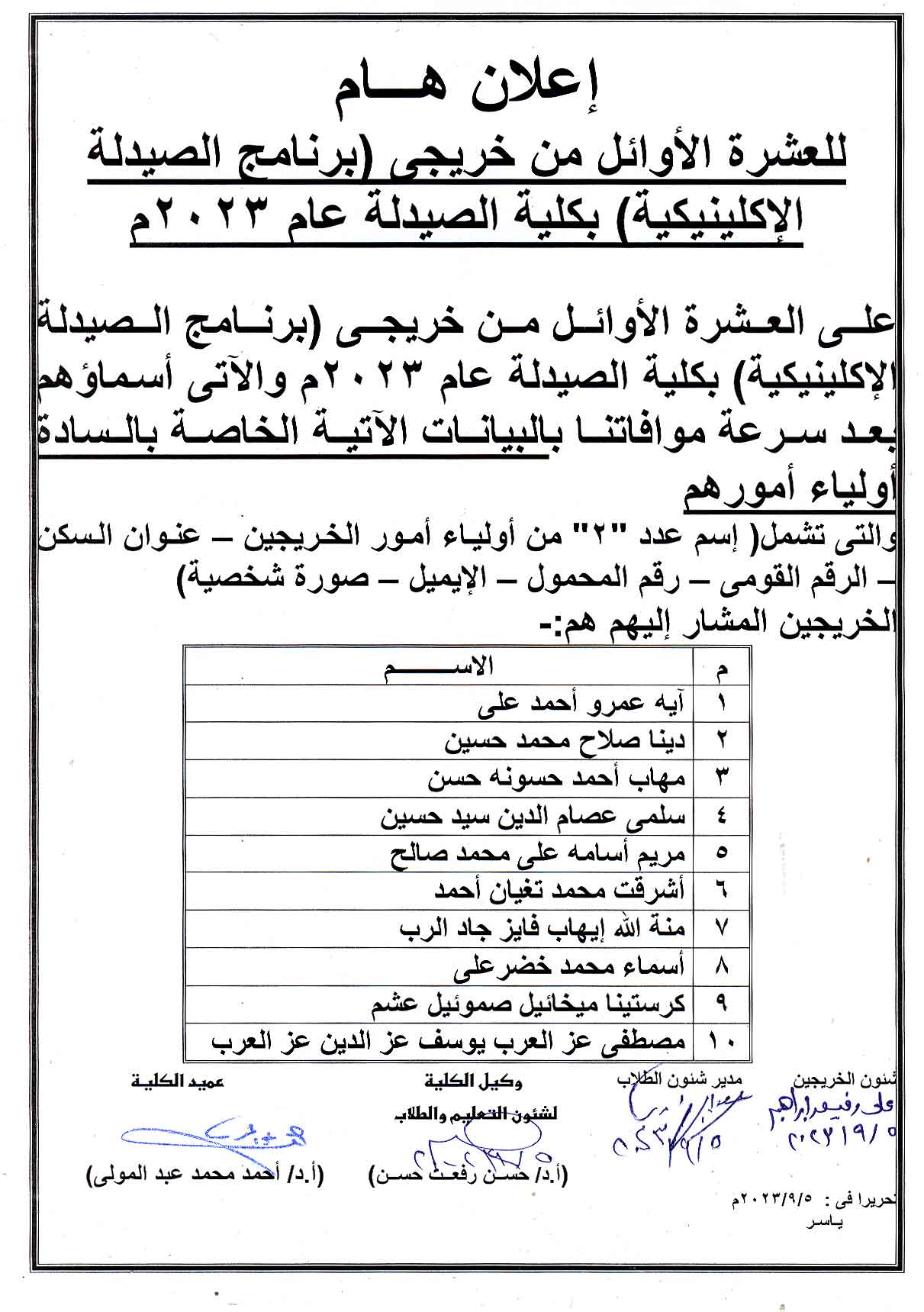
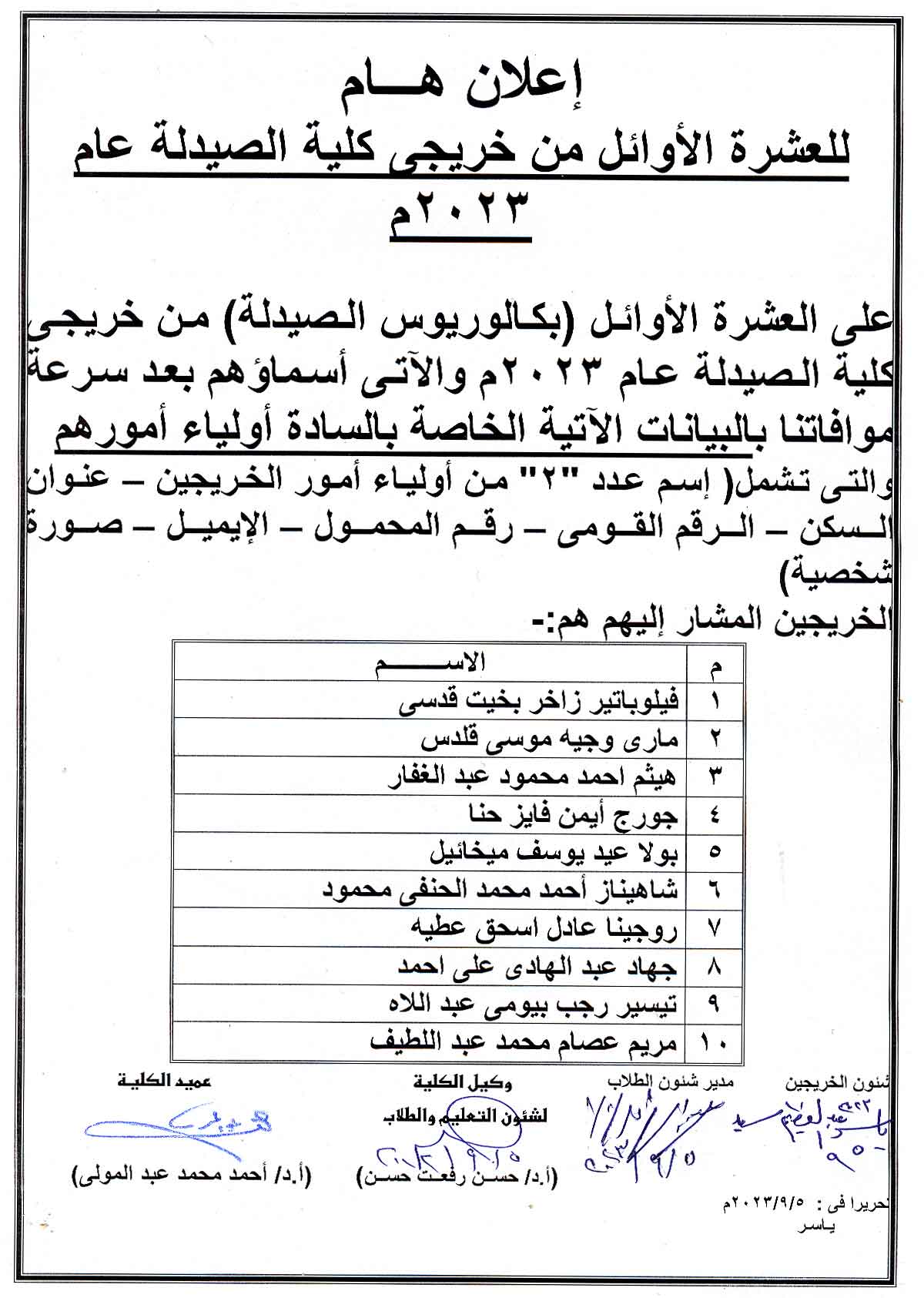
Assiut University announces the research competition for Egyptian university students

تعلن جامعة اسيوط عن المسابقة البحثية ضمن الاحتفالات باليوبيل الذهبى لحرب اكتوبر لطلاب الجامعات المصرية
news category
خبر عام

 Do you have any questions? (088) 2080369 - 2345622 Pharmacy_QAAU@pharm.aun.edu.eg
Do you have any questions? (088) 2080369 - 2345622 Pharmacy_QAAU@pharm.aun.edu.eg
تعلن جامعة اسيوط عن المسابقة البحثية ضمن الاحتفالات باليوبيل الذهبى لحرب اكتوبر لطلاب الجامعات المصرية



God willing, a meeting of the committee for community and environmental development will hold on Monday, September 11, 2023 at 11:00 AM
In the office of Vice Dean for Community Services and Environmental Development Affairs.

God willing, the laboratories and scientific equipment committee will hold its meeting on Monday, September 11, 2023 at twelve (noon)
in the office of Vice Dean for Community Services and Environmental Development Affairs.

(Prof. Ahmed Mohamed Abdel Mawla)
Several infuenza pandemics have impacted our life, each with variable prevalence and severity. In a search for natural
antivirals, further phytochemical investigation of Gardenia latifolia Aiton, Rubiaceae, was conducted. As a result, fve
structurally diverse glycosides were isolated, ofering valuable chemotaxonomic data. Using the crystal violet technique,
three isolates, canthoside C, (6R,7S,8S)-7α-[(β-d-glucopyranosyl) oxy] lyoniresinol, and ecdysanrosin A, were evaluated
for their anti-infuenza A (H1N1) activities. Based on previously reported anti-infammatory activity of the guaiane class,
we investigated the inhibitory efect of (1R,7R,8S,10R)-7,8,11-trihydroxy-guai-4-ene-3-one 8-O-β-d-glucopyranoside, a
rare guaiane sesquiterpene glucoside, on inducible nitric oxide (NO) production by Griess assay. Regarding antiviral assay,
canthoside C was the most active. It considerably inhibited H1N1 infectivity at an IC50 value of 10.93 µg/ml, showing a
selectivity index (SI) of 12.88, compared with acyclovir as a standard. Besides, ecdysanrosin A displayed a moderate selective
antiviral activity with an IC50 value of 28.03 µg/ml. Considering their low cytotoxicity on the host cells, canthoside C and
ecdysanrosin A have additional merit as potential antiviral agents. Despite the claimed anti-infammatory activity of guaianes,
(1R,7R,8S,10R)-7,8,11-trihydroxy-guai-4-ene-3-one 8-O-β-d-glucopyranoside showed a limited anti-infammatory activity